
Within the dynamic discipline of AI know-how, a urgent problem for the drug discovery (DD) group, particularly in structural biology and computational chemistry, is the creation of revolutionary fashions finely tuned for drug design. The core problem lies in precisely and effectively predicting molecular properties essential for understanding protein-ligand interactions and optimizing binding affinities, important for advancing efficient drug improvement initiatives.
In present structural biology and drug design, researchers generally rely on present datasets and strategies, which have inherent limitations like structural inaccuracies, crystallographic artifacts, and difficulties in precisely capturing the dynamic nature of protein-ligand interactions. Conventional approaches for predicting molecular properties typically lack the mandatory element for advanced protein-ligand interactions, neglecting the important position of dynamics and adaptability in understanding binding mechanisms and affinity.
Researchers from the Institute of Structural Biology, Technical College of Munich, Jülich Supercomputing Centre, Helmholtz AI, Cambridge College, Jagiellonian College, and Institute of Computational Biology suggest MISATO, marking a transformative shift in drug discovery and structural biology methodologies. MISATO addresses the restrictions of present strategies by integrating quantum-chemically refined ligand information, molecular dynamics (MD) simulations, and superior AI fashions. This complete strategy facilitates a nuanced understanding of molecular properties, capturing digital construction particulars and dynamic conduct essential for correct predictions.
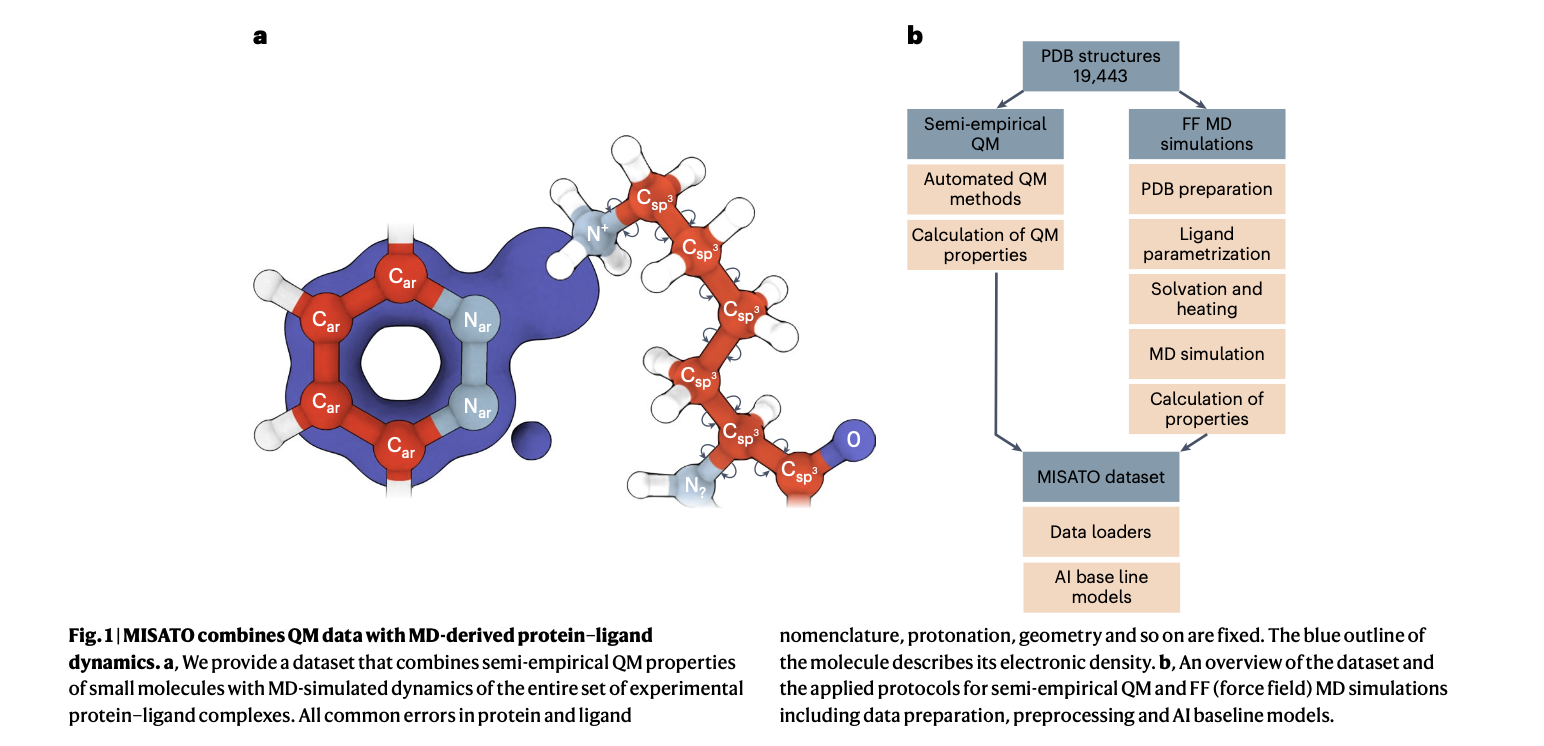
MISATO takes a complete strategy, using semi-empirical quantum chemical strategies to refine ligand datasets. This technique captures digital properties with excessive accuracy, whereas additionally analyzing each digital construction particulars and dynamic conduct, essential for exact predictions. Moreover, classical MD simulations inside MISATO characterize the dynamic conduct and conformational panorama of protein-ligand complexes, providing insights into binding mechanisms and adaptability. AI fashions built-in into MISATO, similar to graph neural networks (GNNs), are educated on this enriched dataset to foretell properties like adaptability, binding affinities, and thermodynamic parameters. Intensive experimental validations affirm the efficacy of those fashions in precisely predicting key molecular properties essential for drug discovery.
In conclusion, MISATO signifies a key stride in AI-driven drug discovery and structural biology. By integrating quantum chemistry, MD simulations, and superior AI fashions, MISATO gives a holistic and strong answer to challenges in structure-based drug design, enhancing accuracy and effectivity and empowering researchers with potent instruments.
Try the Paper. All credit score for this analysis goes to the researchers of this undertaking. Additionally, don’t overlook to comply with us on Twitter. Be a part of our Telegram Channel, Discord Channel, and LinkedIn Group.
When you like our work, you’ll love our publication..
Don’t Neglect to affix our 42k+ ML SubReddit
Aswin AK is a consulting intern at MarkTechPost. He’s pursuing his Twin Diploma on the Indian Institute of Know-how, Kharagpur. He’s enthusiastic about information science and machine studying, bringing a robust educational background and hands-on expertise in fixing real-life cross-domain challenges.